How to Understand Drug Metabolism: A High-Yield Guide for Students
What if you could stop memorizing hundreds of CYP450 interactions and start predicting them instead? Learning how to understand drug metabolism shouldn’t feel like an endless battle against an academic list. You’ve likely spent hours trying to distinguish between Phase I and Phase II reactions or worrying about the clinical impact of the first-pass effect. It’s a struggle many students face, especially as the pharmacology landscape shifts with 79 new active substances launched in 2025 alone and new FDA draft guidance on plausible mechanism pathways emerging in early 2026.
We’re here to help you master the complexities of pharmacokinetics by establishing a clear mental model for drug biotransformation. This guide breaks down the fundamental frameworks of the CYP450 system, turning a daunting list of inhibitors and inducers into a logical chemical tagging process. We’ll explore the core principles you need to identify high-yield drug interactions and secure higher scores on your pharmacology board exams. Whether you’re preparing for clinical rotations or your final certifications, this systematic approach ensures you’re ready for the evolving demands of modern medicine.
Key Takeaways
- Learn how to integrate biotransformation into the broader ADME framework to better predict drug behavior throughout the body.
- Discover the “Functionalization vs. Conjugation” mental model to simplify the distinction between Phase I and Phase II metabolic reactions.
- Master the high-yield CYP450 enzymes and learn how to understand drug metabolism by identifying the “major players” like CYP3A4 and CYP2D6.
- Understand the clinical impact of the first-pass effect and pharmacogenomics to adjust dosing for safety and efficacy.
- Access high-yield study strategies, including mnemonics for the “Big 5” CYP inducers and inhibitors, to prepare for board exam questions.
The ADME Framework: Where Metabolism Fits in Pharmacology
To truly grasp pharmacology, you have to view the body as a complex processing plant. Drug metabolism, or biotransformation, is the chemical alteration of a substance within this system. It’s a critical component of the ADME framework, which stands for Absorption, Distribution, Metabolism, and Excretion. While absorption and distribution focus on getting the drug to its target, metabolism and excretion handle its removal.
The primary objective of this process is to convert lipophilic, or fat-soluble, substances into hydrophilic, or water-soluble, waste. Most medications are designed to be lipophilic so they can easily cross cellular membranes and reach their intended receptors. However, the kidneys struggle to excrete these oily compounds. By chemically modifying them, the body ensures they can be dissolved in urine and safely removed. This Drug Metabolism Overview highlights how these transformations are essential for maintaining physiological balance. While the liver serves as the primary metabolic factory, it’s not the only player. Extrahepatic sites like the kidneys, lungs, and gastrointestinal tract also contribute to the chemical breakdown of medications. Understanding this distribution is a key step in learning how to understand drug metabolism effectively.
Pharmacokinetics vs. Pharmacodynamics
Students often confuse these two pillars. Pharmacokinetics describes what the body does to the drug, while pharmacodynamics focuses on what the drug does to the body. Metabolism sits firmly in the pharmacokinetic camp. Think of it this way: if pharmacodynamics is the action of the medication, metabolism is the exit strategy. It’s the methodical process of winding down that action and preparing for disposal. This distinction is vital for clinical practice, as it helps you predict how long a drug will stay in a patient’s system.
The Concept of Biotransformation
It’s a mistake to assume that metabolism always inactivates a drug. While many reactions do result in inactive metabolites, some drugs are administered as inactive prodrugs that require metabolic activation to work. In other cases, the body creates active metabolites that can prolong a drug’s effect or even cause toxicity. This nuanced view is central to our pharmacology guide principles. Mastering these distinctions is how you move beyond rote memorization and develop a professional clinical intuition. Learning how to understand drug metabolism requires recognizing these chemical shifts as dynamic events rather than static rules.
Phase I vs. Phase II Metabolism: The Two-Step Process
The chemical journey of a drug through the body is best understood as a two-stage process designed to increase water solubility. While every medication follows a unique path, the overarching goal remains consistent: transforming a lipophilic molecule into a polar one that the kidneys can easily recognize and eliminate. Learning how to understand drug metabolism effectively starts with distinguishing between these two distinct phases of biotransformation.
Phase I is often referred to as “Functionalization.” During this stage, the body adds or unmasks a polar functional group, such as a hydroxyl (-OH), amino (-NH2), or carboxyl (-COOH) group. Think of this as adding a “chemical handle” to the drug molecule. Phase II, known as “Conjugation,” involves attaching a large, endogenous polar molecule to that handle. This makes the drug significantly more water-soluble. It is a common misconception that drugs always follow a linear path from Phase I to Phase II. In reality, many medications skip Phase I entirely and go straight to conjugation, but it’s rare for a drug to bypass Phase II and still be excreted efficiently. Detailed insights into these pathways can be found in this resource on the Clinical Pharmacology of Drug Metabolism.
Phase I: Oxidation, Reduction, and Hydrolysis
Oxidation is the most frequent reaction in Phase I metabolism. These reactions are primarily driven by the Cytochrome P450 (CYP450) enzyme superfamily. While Phase I usually prepares a drug for Phase II, it can occasionally produce reactive intermediates. These intermediates are sometimes more chemically active or toxic than the original drug. For example, the metabolism of acetaminophen produces NAPQI, a toxic intermediate that must be quickly neutralized by Phase II pathways to prevent liver damage. If you find these pathways difficult to visualize, practicing with Interactive Pharmacology Flashcards can help reinforce the specific enzymes and reactions involved in each step.
Phase II: Conjugation Reactions
Phase II reactions are the “heavy lifters” of biotransformation. They involve transfer enzymes that catalyze the attachment of polar groups. The most common Phase II reaction is glucuronidation, which uses UDP-glucuronosyltransferases (UGTs) to attach a glucuronic acid molecule. Other vital Phase II reactions include:
- Sulfation: Attaching a sulfate group, often important for steroid hormones.
- Acetylation: Adding an acetyl group, which is highly dependent on a patient’s genetic “fast” or “slow” acetylator status.
- Glutathione Conjugation: A critical defense mechanism for neutralizing reactive intermediates from Phase I.
By the end of Phase II, the drug is typically inactive and highly polar, ensuring it remains in the renal tubule for final excretion.

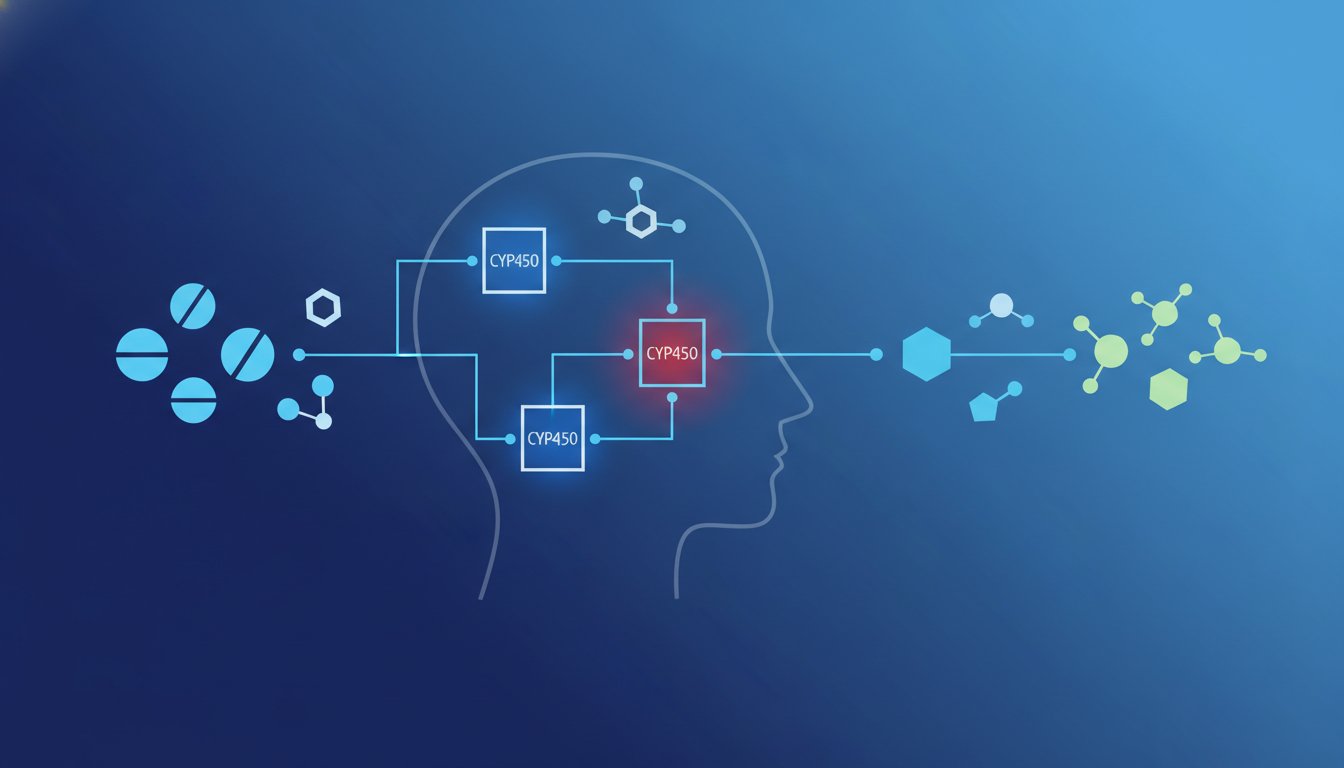
Mastering the Cytochrome P450 System (CYP450)
To master the clinical application of pharmacokinetics, you have to focus your attention on the Cytochrome P450 (CYP450) system. This is a massive superfamily of heme-containing enzymes primarily located in the smooth endoplasmic reticulum of hepatocytes. While the body utilizes dozens of these enzymes, a select group of “major players” handles the vast majority of drug biotransformation. Specifically, CYP3A4, CYP2D6, and CYP2C9 are the high-yield isoforms that appear most frequently on exams and in clinical practice. Learning how to understand drug metabolism at this level requires moving beyond simple definitions and focusing on the dynamic mechanics of induction and inhibition.
Enzyme induction occurs when a substance triggers the body to increase the synthesis of CYP enzymes. This process usually takes several days to weeks to manifest because it requires the transcription of new protein. The result is a faster metabolic rate for any co-administered drugs, which often leads to sub-therapeutic levels and treatment failure. Conversely, enzyme inhibition is much faster, occurring as soon as the inhibitor reaches the enzyme site. Inhibition blocks the enzyme’s activity, causing drug levels to rise dangerously and increasing the risk of toxicity.
CYP450 Inducers vs. Inhibitors
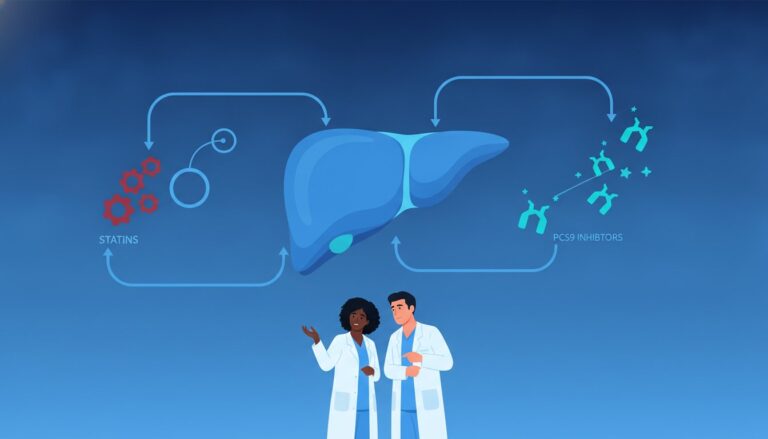
Identifying which substances trigger these changes is essential for patient safety. Common inducers include medications like Rifampin and Carbamazepine, as well as the herbal supplement St. John’s Wort. Patients starting these therapies often need higher doses of their other medications to maintain efficacy. On the other hand, potent inhibitors like Grapefruit juice, Ketoconazole, and Erythromycin can cause drug concentrations to spike. For example, a patient taking a statin who drinks large amounts of grapefruit juice may experience muscle toxicity due to inhibited clearance. For a more comprehensive list of these interactions, you can reference our NAPLEX prep course guide, which categorizes these substances for easier memorization.
Clinical Consequences of CYP Interactions
The impact of these interactions is most severe for drugs with a narrow therapeutic index. Warfarin is a classic case study; because it’s metabolized by CYP2C9, even a slight change in enzyme activity can lead to life-threatening bleeding or dangerous clots. However, don’t forget the “Prodrug” exception, which is a frequent board exam trap. Since prodrugs like Codeine or Clopidogrel require metabolic activation to work, an enzyme inhibitor actually makes them less effective. In these cases, the body can’t convert the inactive drug into its active form, leading to a lack of therapeutic effect. Recognizing these specific patterns is a cornerstone of how to understand drug metabolism in a real-world clinical setting.
Key Factors Influencing Drug Biotransformation
One of the most critical aspects of learning how to understand drug metabolism is recognizing that it isn’t a uniform process across all patients. Biological variables can drastically alter how quickly a drug is cleared from the system. This variability is why a standard dose might be therapeutic for one individual but toxic for another. To provide safe patient care, you must account for the physical and genetic factors that dictate metabolic efficiency.
The first-pass effect is perhaps the most famous example of metabolic influence. When a drug is taken orally, it’s absorbed from the gastrointestinal tract and transported directly to the liver via the portal vein. For drugs with a high first-pass effect, the liver metabolizes a significant portion of the dose before it ever reaches systemic circulation. This explains why an oral dose of a medication is often much higher than its intravenous counterpart, which enters the bloodstream directly and bypasses this initial hepatic processing.
Age and disease also play pivotal roles. Neonates have immature enzyme systems, particularly Phase II pathways, which can lead to dangerously prolonged drug half-lives. Conversely, the elderly often experience a decline in hepatic blood flow and liver mass, reducing their overall metabolic capacity. Pathological conditions like cirrhosis directly damage hepatocytes, while heart failure reduces the delivery of drugs to the liver. Both conditions impair hepatic clearance and require careful dosage adjustments to avoid adverse effects. Understanding these variables is central to how to understand drug metabolism in a clinical setting.
The First-Pass Effect and Bioavailability
Bioavailability is the fraction of an administered dose that reaches the systemic circulation in an unchanged form. Nitroglycerin is a classic example of a drug with nearly 100% first-pass metabolism; if swallowed, the liver destroys it almost entirely. By using sublingual or transdermal routes, clinicians bypass the portal circulation, ensuring the medication enters the bloodstream directly. This practical application is a cornerstone of clinical pharmacology and drug delivery design.
Genetics and Individual Variation
Pharmacogenomics explores how genetic polymorphisms lead to slow versus fast metabolizers. The CYP2D6 enzyme is a high-yield example because it’s responsible for converting codeine into its active form, morphine. Ultra-rapid metabolizers possess multiple copies of the CYP2D6 gene, which can lead to dangerously high morphine levels even with standard codeine doses. This genetic diversity is a primary reason why personalized medicine is becoming the standard of care. You can find detailed breakdowns of these genetic variations in our nursing pharmacology study guide.
Mastering these metabolic nuances is essential for any healthcare professional. If you’re ready to master these clinical nuances, our nursing pharmacology study guide offers high-yield breakdowns of metabolic variations to ensure you are prepared for your next exam.
High-Yield Study Strategies for Drug Metabolism
Rote memorization is often the default approach for many students, but it’s rarely effective for long-term retention. Learning how to understand drug metabolism is less about brute force and more about strategic organization. By using the “Functionalization vs. Conjugation” mental model, you can categorize every reaction into a logical step. Phase I prepares the molecule, and Phase II finishes the job. This structural thinking prevents the confusion that often leads to errors on high-stakes clinical exams. It turns a list of reactions into a predictable chemical narrative.
Beyond the standard pathways, you should focus your attention on the “outlier” drugs. These are medications that bypass the liver entirely or are excreted unchanged in the urine. Recognizing these exceptions early prevents you from applying hepatic rules where they don’t belong. Combining this logic with active recall is the fastest way to build professional competency. Mastering how to understand drug metabolism requires a move toward active learning where you challenge your mental models with real-world scenarios.
Leveraging Mnemonics for Board Success
Board exams frequently test your ability to predict drug-drug interactions. Mnemonics bridge the gap between a list of names and a functional toolkit. For enzyme inducers, use the PS-PORCS acronym: Phenytoin, Smoking, Phenobarbital, Oxcarbazepine, Rifampin, Carbamazepine, and St. John’s Wort. To remember the most common inhibitors, use G-PACMAN: Grapefruit juice, Protease inhibitors, Azole antifungals, Cimetidine, Macrolides, Amiodarone, and Non-DHP Calcium Channel Blockers. Applying these to practice questions helps you quickly identify when a drug level is likely to spike or drop.
Why PharmEDU Beats Traditional Textbooks
Static diagrams in textbooks often fail to illustrate the fluid nature of enzyme kinetics. Our High-Yield Video Vignettes offer a visual narrative that makes these pathways stick in your memory. Interactive flashcards further reinforce this by forcing you to recall the specific enzyme responsible for each reaction. Contrast this approach with a traditional comprehensive pharmacy review to see how digital tools enhance retention. If you want to move beyond the limitations of paper and master these complex systems, Start your PharmEDU subscription today to master metabolism and gain access to our full suite of clinical case studies and pharmacology practice quizzes.
Mastering Clinical Biotransformation for Board Success
Understanding the chemical journey of a drug is more than an academic exercise; it’s a fundamental skill for safe clinical practice. You’ve explored how the ADME framework provides the necessary context for biotransformation and why the distinction between Phase I functionalization and Phase II conjugation is vital for predicting excretion. Learning how to understand drug metabolism allows you to move beyond simple memorization and start anticipating complex drug-drug interactions with professional confidence.
By recognizing how individual factors like genetics, age, and disease states alter drug clearance, you’ve built a robust mental model for your future career. To solidify these concepts and ensure you’re fully prepared for clinical rotations, you can Master pharmacology with PharmEDU’s high-yield study tools. Our platform offers over 100 high-yield video vignettes and interactive flashcards specifically designed for NAPLEX prep. The mobile-compliant micro-learning design ensures you can study effectively whenever your schedule allows, taking the administrative burden out of your professional development. We’re proud to be your partner in this journey, providing the precision and support you need to excel on your exams and in the clinic.
Frequently Asked Questions
What is the difference between Phase I and Phase II metabolism?
Phase I metabolism involves functionalization reactions like oxidation to create a polar handle, while Phase II focuses on conjugation to attach large molecules like glucuronic acid. Most drugs undergo both stages to become water-soluble for renal excretion. Learning these distinctions is a core part of how to understand drug metabolism for clinical exams. This two-step process ensures that lipophilic substances are effectively transformed into waste.
How does the cytochrome P450 system affect drug interactions?
The Cytochrome P450 system dictates drug interactions by serving as the primary metabolic pathway for most medications. When two drugs compete for the same enzyme, or if one drug changes enzyme activity, the concentration of the second drug will shift. This often leads to either therapeutic failure or dangerous toxicity. Mastering these interactions is essential for safe prescribing and predicting patient outcomes in complex treatment plans.
What is a prodrug and how is it metabolized?
A prodrug is a pharmacologically inactive compound that requires metabolic biotransformation to become an active medication. Codeine is a classic example; it has no analgesic effect until the CYP2D6 enzyme converts it into morphine. If a patient’s metabolism is inhibited, the prodrug won’t work. Conversely, ultra-rapid metabolizers might experience toxicity from the sudden spike in active drug levels during this conversion process.
Why is the liver the primary site of drug metabolism?
The liver is the primary site because it possesses the highest concentration of biotransformation enzymes, particularly the CYP450 superfamily. It also receives nearly all blood from the gastrointestinal tract through the portal vein. This anatomical arrangement allows the liver to process and neutralize foreign substances before they reach the rest of the body. While other organs contribute, the liver’s enzymatic density makes it the metabolic factory.
What happens if a drug inhibits a CYP450 enzyme?
Inhibiting a CYP450 enzyme blocks the metabolic pathway for any drug that relies on that specific enzyme for clearance. This causes the drug’s plasma concentration to rise, potentially leading to toxic side effects. For instance, if a patient takes a macrolide antibiotic that inhibits CYP3A4, their levels of co-administered statins could spike. This is a critical concept for anyone learning how to understand drug metabolism and patient safety.
How does age affect how the body metabolizes medications?
Neonates have immature enzyme systems, particularly Phase II pathways, which significantly slows their ability to clear certain medications. In contrast, elderly patients often experience reduced hepatic blood flow and a decrease in total liver mass. Both extremes of age result in a diminished metabolic capacity compared to healthy adults. Clinicians must adjust dosages for these populations to prevent drug accumulation and adverse reactions during therapy.
What is the first-pass effect in pharmacology?
The first-pass effect refers to the rapid hepatic metabolism of an orally administered drug before it reaches the systemic circulation. After absorption in the gut, the drug travels through the portal vein directly to the liver. If the liver clears a large percentage of the dose during this initial pass, the drug’s bioavailability is greatly reduced. This explains why oral doses are often much higher than intravenous doses.
Which drugs are known as potent CYP450 inducers?
Potent CYP450 inducers include medications such as Rifampin, Carbamazepine, Phenytoin, and Phenobarbital. These substances trigger the body to synthesize more metabolic enzymes over a period of days or weeks. This increase in enzyme count speeds up the breakdown of other drugs, which often results in sub-therapeutic blood levels. Students often use the PS-PORCS mnemonic to remember these high-yield inducers for their pharmacology board exams.